IVDR — In Vitro Diagnostic Regulation — is the new European regulation 2017/746, relating to in vitro diagnostic medical devices (IVD), which entered into force in May 2022. It is intended to guarantee their greater effectiveness and to harmonize the rules in the EU. At the same time, it increases safety levels for patients. Let’s take a look at this new regulation. What type of material is affected? What are the changes? And how can in vitro diagnostic manufacturers and operators be supported to comply with regulations on the European market?
What is a DM – DIV?
Let’s start by clarifying what a DM-IVD or “In Vitro Diagnostic Medical Device” is. The definition is broad, it corresponds to any element designed by the manufacturer for the in vitro analysis of human biological matrices (urine, blood, tissue, etc.):
- reactant or reactive product;
- sample container;
- instrument, apparatus, equipment;
- calibration device;
- software or any system.
The analysis is carried out with the aim of providing different types of information:
- on a physiological or pathological process or state;
- on physical or mental birth defects;
- on predisposition to pathology or disease;
It may also concern other applications, such as:
- determining safety and compatibility with potential recipients;
- predicting a response or reaction to treatment;
- the definition or control of therapeutic measures.
IVDR: what is the purpose of the new European regulations on DM-IVDs?
The IVDR repeals Directive 98/79/EC on in vitro diagnostics IVDD (In Vitro Diagnostic Devices Directive) 98/97/EC. After a transition period which lasted 5 years, the new regulation came into force on May 26, 2022. Since this date, any in vitro diagnostic device placed on the European market must therefore comply with this regulation.
It makes it possible to meet increased security requirements in the field of medical diagnostics. In addition, the regulation does not need to be transposed into the national laws of EU member states, which limits the risks of differences in interpretation in different countries. Thus, the IVDR also facilitates European harmonization, with optimal governance of the medical device sector within the EU. On the other hand, in better correspondence with technological developments, it will make it possible to better adapt to innovation and upgrade the image of medical device brands.
New European regulation on in vitro diagnostic devices: what’s changing
Overall, the new European Regulation EU 2017/746 (IVDR) has redefined the classes of In Vitro Diagnostic Devices (IVDs), as well as the evidence of their analytical performance. The analyzes are more exhaustive and more demanding. Using an EN ISO/IEC17025:2017 accredited laboratory can enhance the reliability and accuracy of the data. It will therefore facilitate the transition to IVDR, with better guarantees of compliance and appropriate support.
Here are the main areas of change introduced by the IVDR.
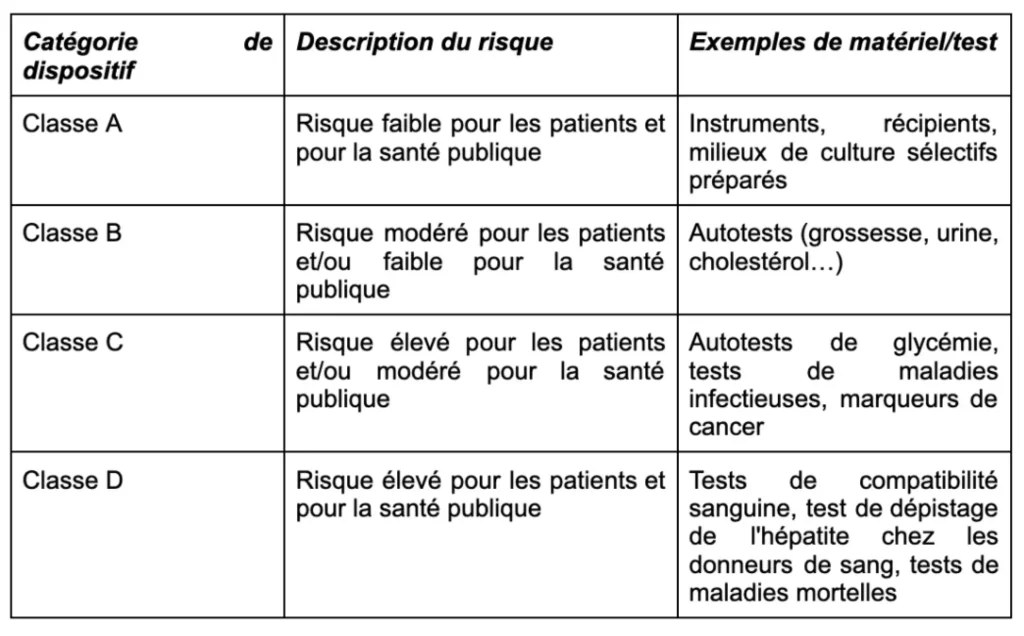
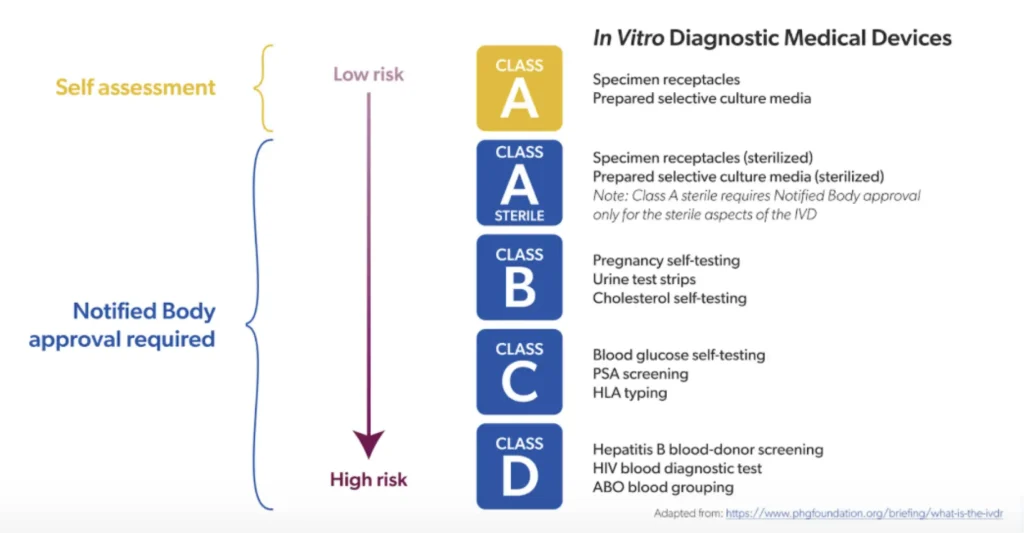
Classification of diagnostic devices according to risk level
The old directive did not require independent assessment for different types of devices. The IVDR has defined 4 classes (A to D), from the lowest level of risk to the highest.
More rigorous clinical evidence of device performance and safety
Based on the performance evaluation report, diagnostic tests must be supported by clinical demonstrations and demanding technical information.
Conformity assessment of high-risk devices by notified bodies
The evaluation of class B, C, and D devices must be entrusted to a notified body. It is more rigorous than before, and varies depending on the material. For class D devices, additional assessment by a European Union reference laboratory is also required.
Reinforced surveillance
Manufacturers are subject to monitoring by Notified Bodies (NBs), which themselves are controlled by EU member states. Pharmacovigilance requirements are also more stringent. The evaluation of device performance must, moreover, be carried out post-marketing and throughout the life of the product.
Increased transparency and traceability
Information about the devices and their performance is public. They are available in a new European database on medical devices (Eudamed). Incident notification times have been extended and incident reports are integrated into the database.
Responsibility extended to all operators
Responsibility for the conformity of medical devices is explicitly assigned to all economic players in in vitro diagnostics:
- manufacturers;
- authorized representatives;
- importers and distributors of in vitro diagnostic devices in the European Union;
- regulatory affairs or quality management professional involved in IVD.
How to be well supported towards European regulation EU 2017/746?
The changes induced by the IVDR not only involve specialized and additional expertise, but also a significant workload and costs. According to Euractiv, in December 2023, 80% of in vitro diagnostic devices were subject to control by a notified body. This corresponds to approximately 24,000 devices!
Good support in monitoring the performance and safety of IVDs is therefore crucial for companies in the medical device sector. Compliance with EU regulation 2017/746 is essential. And faced with the shortage of notified bodies, highlighted by the In Vitro Diagnostic Industry Union (SIDIV), the risks of delay in marketing are significant for those who have not found a reliable and compliant partner.
- The keys to a successful transition to IVDR are:
- Hire a laboratory specializing in the analytical performance of in vitro diagnostic devices. ISO/IEC17025 accredited for this activity, it will allow you to better guarantee regulatory compliance.
- Get help managing quality management system plans and reports.
- Carry out a gap analysis: this process consists of comparing the current situation with the projected situation, with the aim of identifying the strategies necessary to close the identified gaps.
- Register on the European EUDAMED database.
- Get support for post-marketing surveillance and material vigilance.
Amarok Biotechnologies, trusted ISO/IEC17025 accredited partner, for evaluating the performance of DM-IVDs
In France, Amarok Biotechnologies is the first EN ISO/IEC17025:2017 accredited laboratory for the evaluation of the analytical performance of in vitro diagnostic medical devices. This recognition by the Human Health division of COFRAC allows it to offer its customers the best guarantees of compliance with the new IVDR regulations. Added to this is its expertise in data generation and performance testing for medical diagnostic automation systems and reagents since 2011.
The implementation of a rigorous quality policy has also been at the heart of the company since its creation. Its analytical performance evaluation laboratory, as well as all of its activities, are ISO 9001:2015 certified.
The laboratory is also involved in consulting, expertise and training missions for its various branches, including R&D. This makes the company a partner of choice for support and the preparation of regulatory files.